
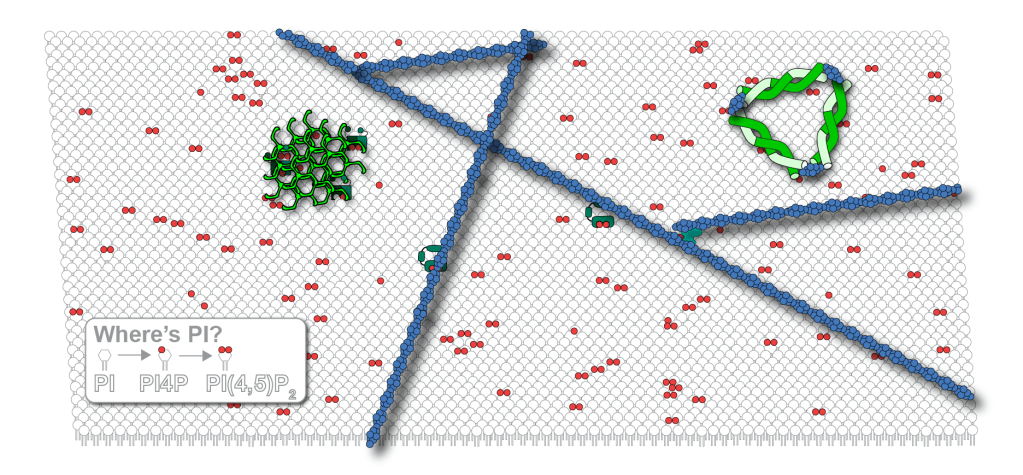
As you may have read elsewhere on this site, the lab studies all the different cellular processes regulated by a very powerful lipid molecule found in the cell membrane, PIP2. PIP2 (and other so-called phosphoinositides, or PPIns) are made from a parent lipid, PI (that stands for Phosphatidylinositol if we want to be all biochemically). Whereas the PPIn are found in very low abundance in cells, PI is actually one of four major membrane phospholipids in our cells; about one in ten lipids are PI. For that reason, we’d always sort of ignored it. We figured there was so much around in cells, PI availability wouldn’t affect PPIn synthesis and so wouldn’t impact cellular physiology much.
However, nobody had really tested this in an intact, living cell before. The main reason is difficulty: the other PPIn all regulate specific proteins, which recognize the lipids very selectively. We can therefore hijack these proteins to make fluorescent biosensors and light up the PPIn distribution in cells (see our 2019 JCB paper for an example). Not so for PI. Because it’s more of as structural lipid, it doesn’t seem to have lipid binding proteins that we can engineer like that.
Instead, we can use a neat trick employed for other lipids in the 80s and 90s. Take some serum albumin and load it with fluorescent lipid (PI in our case). The albumin delivers the lipid to the outer cell membrane, where it piles up. As this happens, some gets taken-up by the cell and transported like the native lipid. We then backwash the excess off the outer membrane and hey-presto! See what lights up:

To our surprise, lots of internal organelle membranes became loaded with PI, but the cell membrane really didn’t. This might indicate the cells don’t leave much PI at their membrane. But if you were a peer-reviewer (hopefully you’re not!), you might argue – perhaps the cell handles fluorescent lipids differently? Fair point actually; the cell transports some fluorescently-labeled lipids normally, but not others. Can we be sure it gets PI right? Not really. We needed more experiments.
So we hadn’t thought of a way to detect PI. But in the lab, we did have a biosensor to detect a PPIn, PI4P. This is made in one step from PI by an enzyme, PI4K. So, we engineered PI4K trigger is rapid recruitment onto different membranes, and measure if the enzyme could could make PI4P there. One example was mitochondria:
Yup, lots of PI to convert to PI4P there! In fact, we found at least some PI4P in most membranes – except the cell membrane. Sounding convincing? Well, put your reviewer hat on, and doubt everything: Maybe the cell membrane has enzyme activities that degrade the PI4P. Actually, that’s another fair point. They do. Hmm, we needed yet another experiment.
So, what don’t cells normally convert PI into? Well, although our cells have a lipid digesting enzyme called phospholipase C (or PLC), this is selective for PIP2. We don’t have anything that normally chews up PI. However, some bacteria pathogens (like Listeria, which causes listeriosis – nasty) have their own PLC. They use these to remove protective glycoproteins that coat the outside of the cell membrane, protecting it. As a side product, they leave behind the fatty lipid tail, known as diacylycerol. Well guess what? We have a great biosensor for diacylglycerol. So, we can take the Listeria PLC gene, activate it inside cells and read-out where there was some PI to convert to diacylglycerol.
Except there’s a problem here. Listeria PLC is very active. And destroying one in ten membrane lipids inside a cell is not exactly good for it. Take one in 10 bricks out a wall – it might still keep standing, but good luck keeping the wind and rain out!
So, we needed to do these experiments, quickly – activate the enzyme in the cell in one go, and then see straight away where the diacylglycerol appears, before the organelle or cell is too badly damaged. To do that, we split the gene in two, and expressed both halves in the cell. We can could then use chemical genetics to stick the resulting protein halves back together by adding a drug called rapamycin, which glues another two proteins together. Attaching these glue-able proteins to the two halves of PLC reconstitutes the active enzyme on demand! Here’s an example, again attaching one half of the PLC to the mitochondria:
Wollap! Again, obviously lots of PI there. In fact, we found evidence of PI in most organelle membranes. But not the inside surface cell membrane, where PI4P and PIP2 are found. We were convinced. Two reviewers were convinced. Well, sort of.
They say great minds think alike. Well, so does Gerry and his former mentor, Tamas Balla. Tamas’ lab used a similar approach. They engineered another PLC from Bacillus (engineered a bit differently) and found the same thing. They also mutated the PLC gene so it would bind to PI, but not hydrolyze it (D’oh, why didn’t we think of that? Well, actually it doesn’t work with the Listeria enzyme for some reason). Again, they saw this biosensor in lots of organelle membranes, but not the cell membrane:

Why is that a big deal? Well, it means that if a cell is going to make enough PIP2 to keep its physiology ticking over nicely, supply of PI from these there organelles must be a crucial regulatory step. And we don’t understand very much about how this works currently. We don’t know exactly what regulates it, and whether it goes wrong in disease. That said, we do know everything from infectious diseases to cancer and congenital disorders such as muscular dystrophy can be associated with failure to control PIP2 levels. So, we better figure out how PI traffic to the membrane works – it might reveal other diseases where PIP2 levels are disrupted, and might give us clues to restoring PIP2 in the diseases we know about.
Now, if you go back to the picture at the top of the page, you will understand that there isn’t a mistake in the picture. There really is a representative amount of PI that cartoon of the cell membrane. Can you find it?
To read more about this work, you can link to our paper and Tamas’ here. These were published back-to-back, though ours was accepted a day earlier, so we’re taking all the credit for the discovery 😉
