
This paper represents our first foray into a very important lipid signaling pathway in cancer. Specifically, this is the PI3K pathway -standing for “PhosphoInositide-3-kinase”, an enzyme that add phosphates to phosphoinositide lipids at the 3-position of their head groups.
You can read the full paper for free online at the Journal of Cell Biology.
So, PI3K’s main job is to produce a lipid signal as soon as a growth factor activates a receptor at the cell surface. The lipid signal is recognized by a set of proteins inside the cell that activate growth and proliferation programs, causing the tissue to start growing:
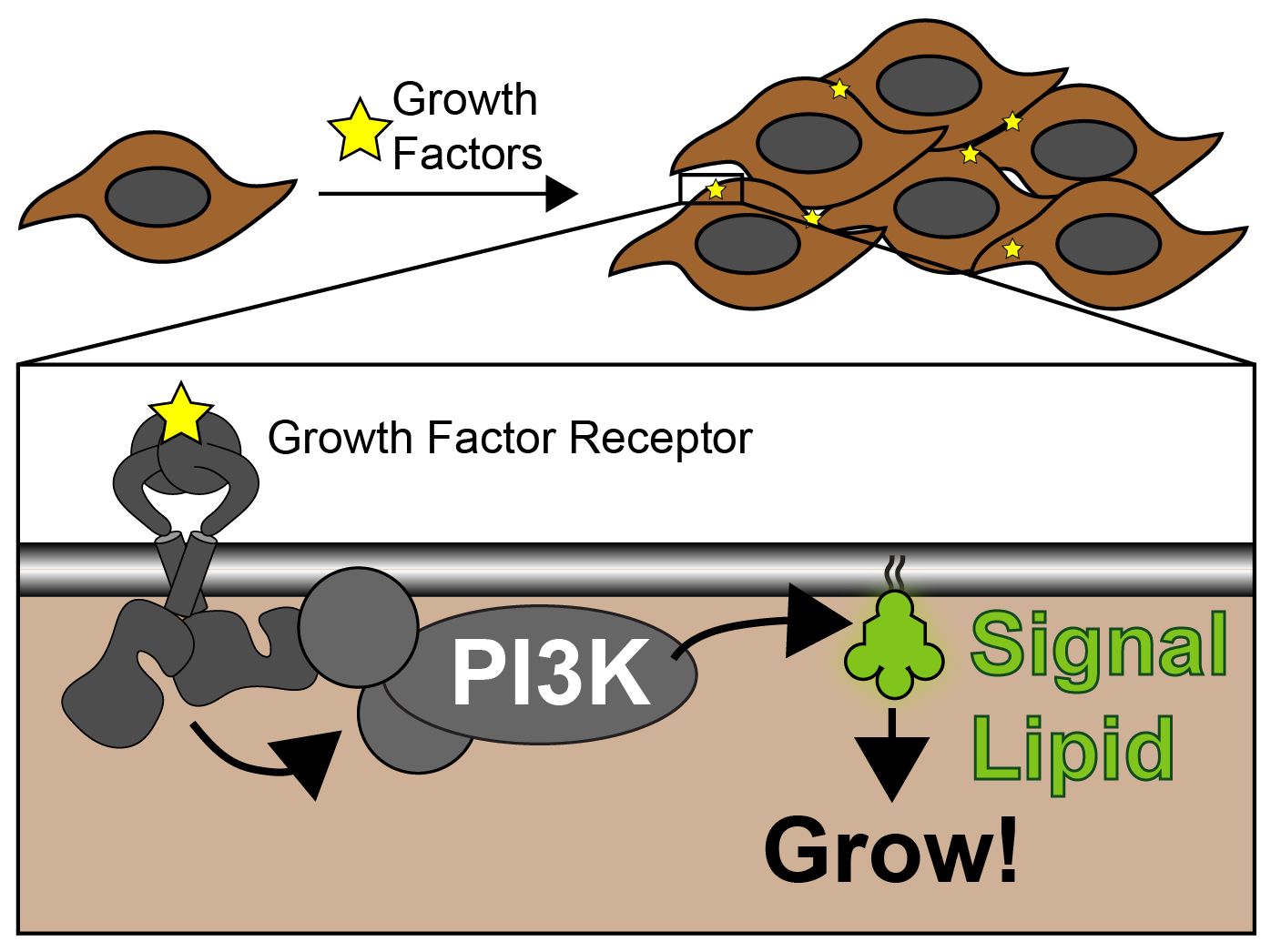
As you can imagine, if PI3K were to become active and start making signal lipids without the activating growth factors, we would get uncontrolled tissue growth. In fact, this is what happens in more than half of cancers.
There are currently a lot of drugs in cancer trials that block PI3K. However, while these can be effective at slowing cancer growth, they can also stop regular functions of PI3K in the body. For example, the body uses the same lipid signals inside cells to stimulate glucose uptake in response to insulin. Obviously, we want processes like that to continue normally. So how can be differentiate the cancer signaling?
As a first step, we decided to take a closer look at the signal lipids that PI3K produces. There are actually two of these; PIP3 and PI(3,4)P2:
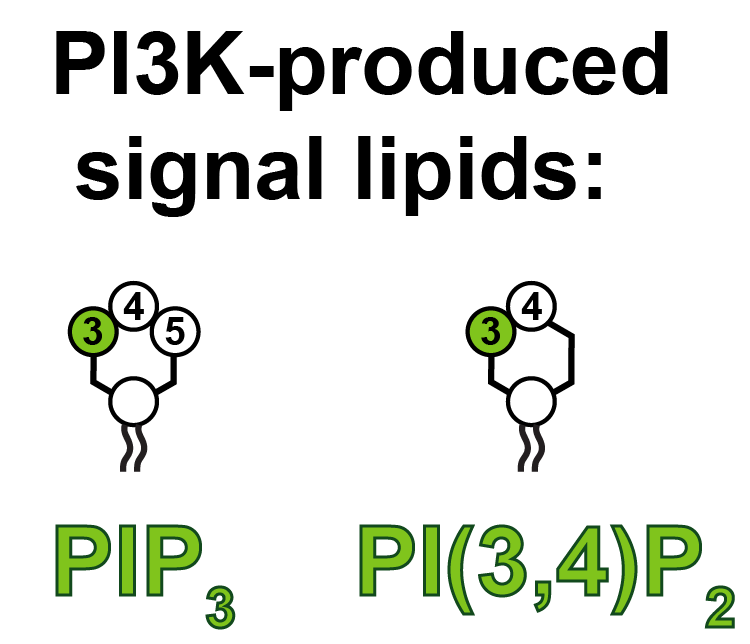
Historically, we’ve known the most about how PIP3 operates; PI(3,4)P2 does largely the same things, but is produced in smaller quantities – it’s been thought of as a sort of molecular side-kick. But is that really all it does? We decided we should come up with some precise and sensitive tools to detect this lipid in living cells, so we can investigate.
To that end, we engineered a specific protein sequence from a protein that sticks very selectively to PI(3,4)P2. We can attach a fluorescent protein to the lipid interacting part (a “PH domain” to use the jargon) so we can see where the in what cell membranes the lipid is being made. However, because there’s so little PI(3,4)P2, it’s still hard to see. To try and boost the signal, we stuck three lipid interacting proteins together:
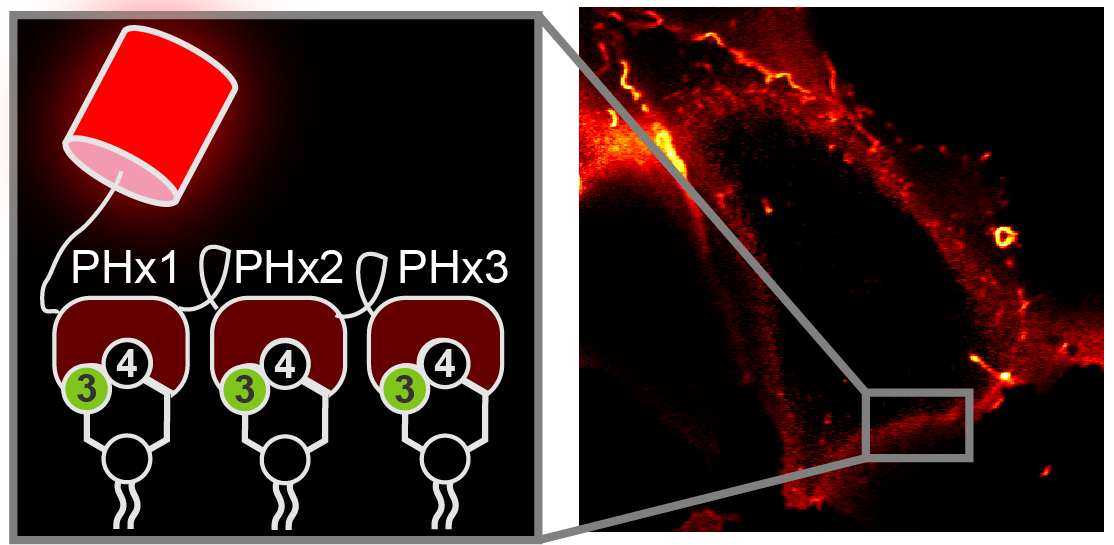
That looked pretty promising: we could clearly make out the “PHx3” probe at the cell membrane.
Is this really because PI(3,4)P2 is found there? That was a crucial question to answer. In the paper, we did a lot of experiments with neat chemical genetic tricks to show that removing PI(3,4)P2 – and not any other lipids – prevented the PHx3 going to the cell membrane.
To be certain, we also wanted to show that PI(3,4)P2 was all that the PHx3 probe needed to go to a membrane. To that end, we collaborated with a team from the Chemistry department to engineer a bacterial enzyme: this enzyme makes PI(3,4)P2 in vesicles inside the cell, where the lipid isn’t normally found. We incorporated an unnatural amino acid into the enzyme with some very clever genetic trickery; that amino acid stops the enzyme producing PI(3,4)P2 right up until we shine violet light on the cells. That light converts the unnatural amino acid to naturally occurring lysine. With that, the enzyme springs to life, filling endosomal vesicles with PI(3,4)P2. As you can see, that massively pulls our PHx3 probe to the vesicles:
This made us very happy, because it showed we now had a very selective, sensitive probe with which to detect PI(3,4)P2 in living cells. So what now? We figured we could now answer some questions about PI(3,4)P2’s relationship with its bigger and more famous brother, PIP3.
PI3K is known to make both lipids. The simplest route is direct: PI3K adds a phosphate to the lipids PI(4,5)P2 or PI4P to make PIP3 and PI(3,4)P2, respectively. However, we also know that cells can convert PIP3 to PI(3,4)P2 with an enzyme that pulls a phosphates at the 5-position off – i.e., PI(3,4)P2 is made via an indirect mechanism:

Do either or both of these pathways operate in cells after PI3K activation? To answer this question, we figured that if we depleted PI(4,5)P2 from the cells, they would not be able to make PIP3 – leaving only the “direct synthesis” pathway available to the cells:
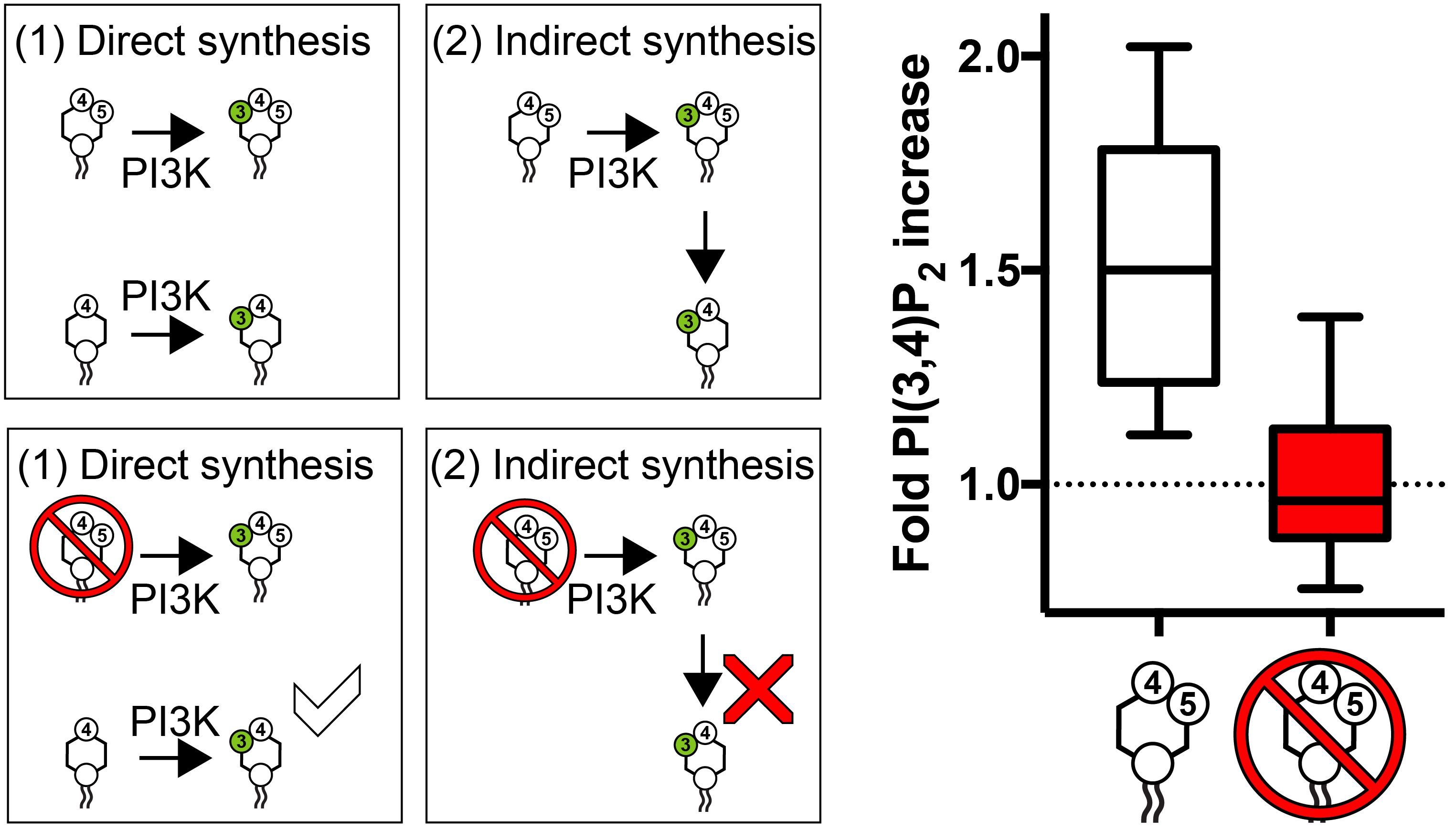
As you can see from the graph, when PI(4,5)P2 is gone, the cells cannot accumulate PI(3,4)P2. QED, they have two use the “indirect” synthesis pathway to make PI(3,4)P2. Thats valuable information, as we now know that we could possibly interfere with the conversion step, blocking PI(3,4)P2 production without affecting PIP3.
A second question we wanted to know was how cells get rid of these signal lipids once they’ve done their job. An enzyme called PTEN is known to break down PIP3, converting it back to PI(4,5)P2. Actually, PTEN is a tumor suppressor – can you guess why? It turns out that very often, cancer cells lose their PTEN enzymes – causing PIP3 to accumulate, meaning they can’t switch off the growth signal.
So, given that PI(3,4)P2 comes from PIP3, of course we will accumulate both lipids when PTEN is lost. But what about in normal cells – how is PI(3,4)P2 removed to help switch off the signal? A different enzyme, INPP4, was known to operate here. But some of our colleagues from Cambridge suggested that PTEN might also remove PI(3,4)P2:

Does this really happen? That’s a tough ask – because when PTEN removes PIP3, it will also stop conversion to PI(3,4)P2, so both lipids will disappear. That’s quite different from PTEN directly removing PI(3,4)P2. How can we distinguish these?
Well, you might remember that one of our earlier experiments used a bacterial enzyme to make PI(3,4)P2 on vesicles inside the cell. Unlike at the cell membrane, this PI(3,4)P2 is made directly, and there is no PIP3. So, having made this vesicular PI(3,4)P2, we could now use a chemical-genetic trick to switch on PTEN – and see if it removes our PHx3 probe from the vesicle, demonstrating PI(3,4)P2 breakdown:
Hey presto! It does. So, PTEN not only breaks down PIP3, it also breaks down PI(3,4)P2.
OK… what have we learned from all of this? Well, we saw that PIP3 gets converted to PI(3,4)P2. We also saw an important tumor suppressor gene, PTEN, is involved in removing both lipids. The implication now is that PI(3,4)P2 might have much more than a side-kick role in growth signaling after all. In fact, PI(3,4)P2 seems to hang around at the membrane quite a bit longer than PIP3.
The lab is now investigating whether PI(3,4)P2 is actually a big driver of growth signaling in cancer. If it has an outsize role relative to PIP3, that could be important, since we might have an opportunity to selectively target tumor growth, whilst leaving PIP3-driven insulin signaling relatively unscathed. Work is ongoing!
In the meantime, you can read the full paper for free online at the Journal of Cell Biology.
